






藥品微生物限度檢查(以下簡稱微限)的目的是確定藥品(含原料及輔料)是否汙染微生物或其汙染微生物的程度,將微生物的種類和數量限製在一定的範圍之內或者不得檢出,是保證用藥的有效性、保障藥品安全性的重要措施。也是衡量藥品生產,流通以及使用全過程微生物控製水平的主要依據之一。科學技術的進步,藥品生產工藝的提高以及人民對藥品安全理念的不斷提升,促使藥品檢驗標準不斷提高,微生物限度檢查作為藥品質量的安全性評價指標也不例外。我國的首版藥典於1953年出版,但開展藥品微生物汙染檢查工作始於1972年,《中國藥典》(1995年版)初次收載了微生物限度檢查法,2000年版首次收載了微生物限度標準,2015年版形成了較為完善的藥品微生物控製標準體係,並解決了基本與美國藥典等國際人用藥品注冊技術要求國際協調會(ICH)方案接軌。《中國藥典》(2020年版)進一步促進了藥品微生物實驗室的管理與國際接軌,實現了微限檢查由最終產品檢驗向過程控製的改變。為了提升藥品微生物實驗室的檢驗能力,選取了2019年1月至2020年12月期間實驗室微限檢查結果作為樣本,對微限檢查誤差來源進行回顧性梳理,以期減少或避免誤差出現的概率,保證微限檢查結果的準確性。
1 資料與方法
1.1 資料
選取2019年1月-2020年12月,本實驗室承擔的藥品微限檢查共870批,其中片劑372批、丸劑187批、顆粒劑227批、口服液84批。對研究藥品微限檢查誤差的相關文獻進行檢索,並分類整理。
1.2 方法
按照標準對870批藥品進行微限檢查,記錄發生誤差的情況,分析誤差產生的來源並對檢驗中出現的誤差進行統計,采用Microsoft Office Excel 2007進行整理分析,以回顧性分析的方法探究出現誤差的影響因素,了解其分布情況。同時對比文獻研究數據,觀察本實驗室與其他地市級藥品檢驗機構的差異,以期在未來的工作中減少誤差發生率。
2 結果
對上述870批藥品微限檢查的統計結果顯示,有42批次(4.8%)因受到操作環境、供試液製備等因素影響導致檢驗結果出現偏差。影響因素、誤差例數及占比情況見表1。
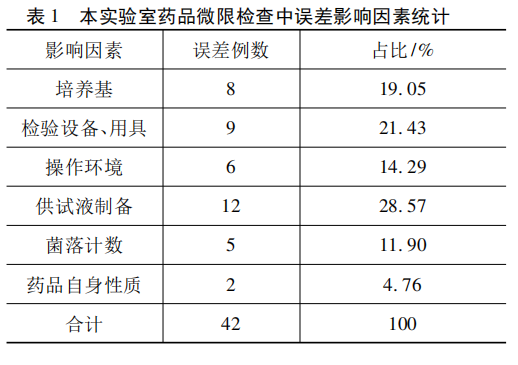
通過中國知網數據庫查閱文獻資料,選取部分高同質性的藥品檢驗機構微生物實驗室的文獻資料,研究統計並進行綜合分析,共1180例誤差數據,詳見表2。
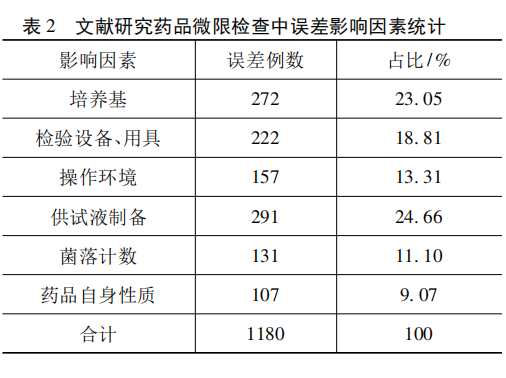
表1和表2對比情況見圖1。可以看出,藥品微限檢查誤差中,影響因素主要來自六個方麵,其中供試液製備時出現誤差比例最高,其次是培養基,另外依次是檢驗設備及用具、操作環境、菌落計數和藥品自身性質。本實驗室和其他地市實驗室統計結果基本一致。
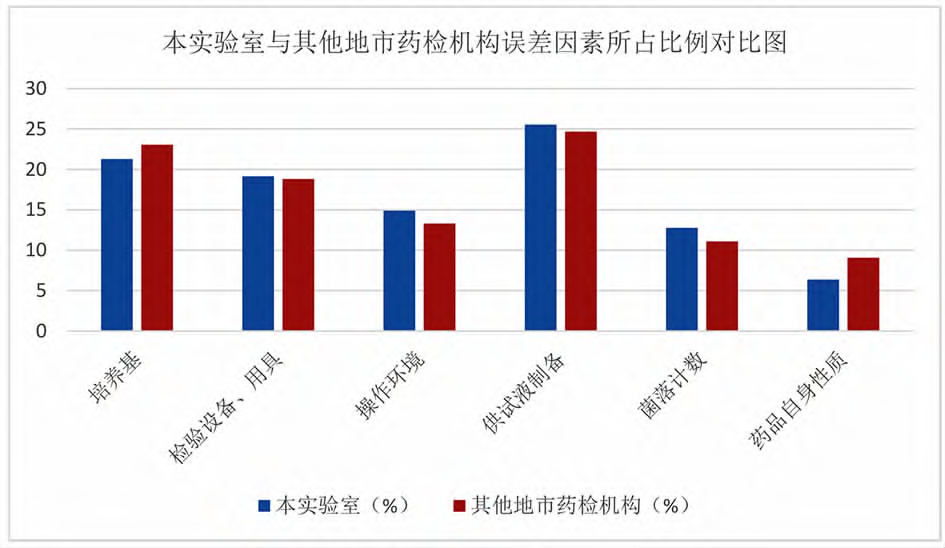
圖1本實驗室與其他地市藥檢機構誤差影響因素所占比例對比圖
3 誤差影響因素的分析及探討
藥品微限檢查目前最常用的方法是生物學檢驗技術,該技術操作程序較為繁瑣、檢驗流程要求緊密嚴格,檢驗周期較長,導致出現幹擾因素的環節較多,容易導致檢驗結果出現誤差。因此,總結分析誤差影響因素,在今後的檢驗工作中給予充分關注,可以減少誤差出現的概率,保證檢驗結果的準確性與可靠性。
3.1 供試液製備
供試液製備是誤差發生的主要因素,該工作目前主要由檢驗人員完成,沒有自動化儀器設備可以替代。因此,檢驗人員的專業素養、理論知識、操作技能和工作經驗是嚴格規範操作、保證結果準確性的基礎。
供試液製備時應保證樣品的真實汙染狀態,既不能殺死樣品本身汙染的微生物,也不能汙染樣品,還要避免操作不當導致樣品汙染實驗環境。要通過方法適用性檢查,保證供試液不具有或者消除其殺菌及抑菌活性。在注皿之前,應保證樣品分散均勻,避免吸取供試液的沉澱物或上清液造成結果偏離。為防止細菌增殖,製備過程要穩、準、快,從供試液製備到傾注培養基整個操作過程應嚴格控製在1小時內,避免時間過長對檢驗結果造成影響。
鑒於藥檢機構成立時間早,人員更新慢,專業結構不合理,人員老化較為嚴重,現有工作人員大部分經曆了多版藥典的實施。有些人員學習能力較差,學習意識淡泊,不能及時跟進檢驗標準的變化,不能有效理解新版藥典微限檢查的操作要求,容易在操作中出現失誤,最終導致出現錯誤的結果。主要表現在以下幾個方麵:一是有些藥檢機構認為微限檢查不使用大型精密儀器,檢驗方法較為單一,對人員和技術要求不高。在人員配置時將非微生物學專業畢業的人員、無相關微限檢查工作經曆的人員安排到微限檢查崗位; 二是藥檢機構中相比較於理化檢驗人員,微限檢查人員較少,考慮到培訓成本,“請進來”的培訓幾乎沒有。除了技術骨幹極少的外出培訓機會,大多靠檢驗人員自學,學習的自覺性、學習的效果均難以追蹤考核; 三是上崗考核流於形式。對於新進人員或者轉崗人員,上崗考試基本以筆試為主,形式大於內容,對操作技能重視不夠。
3.2 培養基
培養基是微生物檢驗的基礎,直接影響微生物檢驗結果。目前,大部分實驗室使用商品化培養基,收到培養基後應對品名、批號、生產廠家及有效期等進行初步驗收,使用前應進行適用性檢查,適用性檢查符合要求方可使用。開瓶時應注明開瓶日期和經驗證的有效期,一旦發現受潮結塊等現象,應立即停止使用。使用時應按標簽標識的說明進行配製,配製時應充分溶解,一般采用濕熱滅菌法。為保證滅菌時既不破壞有效成分,又要保證滅菌徹底,需按照經驗證的滅菌條件操作。應確定每批培養基滅菌後的pH值的範圍(冷卻至25℃左右測定),溫度過高測定pH值時顏色反應可能有誤差。滅菌後的培養基pH值如果測定時超出範圍,不便調整。因此,建議實驗室在滅菌前測定培養基pH值,一旦超出範圍,應緩慢滴加適宜的堿液或者酸液進行調整,添加過量導致再矯正時,培養基的靈敏性可能降低,影響菌落生長。
陳曉燕指出滅菌後的培養基可以在2~8℃放置60天,但因為各實驗室溫度、相對濕度、冰箱性能等控製條件有差異,如要延長保存時間,需要在本實驗室開展驗證。因此,建議臨用新製,避免培養基保存期間導致的促生長能力、抑製能力及指示特性下降。
3.3 檢驗設備及用具
儀器設備應定期進行檢定/校準,不得使用超過檢定/校準周期的儀器設備。儀器設備運行期間,應嚴格按照維護計劃執行,以保持良好的工作狀態。影響實驗結果準確性的關鍵設備有高壓滅菌器、冰箱以及培養箱,在運行過程中對溫度、壓力等關鍵參數要連續觀測,並做好記錄,尤其是高壓鍋,遇到儀器出現故障時要及時發現並處置,避免出現意外傷害事故。本實驗室曾有一台高壓滅菌器在達到滅菌條件和滅菌時間後,仍然保持高溫高壓,為防止意外,隻能強製斷電中止設備運行,本次滅菌的培養基作廢棄物處理。
檢驗中用到的玻璃儀器等用具均需進行滅菌,目前大部分實驗室改用一次性無菌培養皿,省去了培養皿清潔和滅菌的繁瑣。但使用一次性無菌培養皿前,尤其在更換生產廠家後,應注意驗收,可選用少量與庫存培養皿進行對比試驗。本實驗室曾在某次實驗時出現多批不同品種樣品菌落數超標,同時陰性對照有菌落生長,當次實驗結果全部作廢。啟動偏差調查,經回顧性分析和反複討論,考慮是新到貨培養皿的問題。使用原庫存的培養皿進行對比驗證後,確認是培養皿不合格,隨即聯係供應部門對該批次培養皿進行了退換,同時建議供應部門加強對供應商的評估。
3.4 操作環境
為降低或避免交叉汙染,微限檢查應在不低於D級背景下的生物安全櫃或B級潔淨區域內進行,且操作區域內應為單向流空氣。為保持環境的適宜性和有效性,年初應製定環境監測計劃,定期監測浮遊菌、沉降菌、壓差等重要參數。空調潔淨係統長時間運行後,初效、高效過濾網更換不及時,會有潔淨度不符合要求的風險。檢驗人員應具有無菌意識,將無菌觀念銘記在心,落實在細節處。
檢驗前要做好充分的準備工作,將所需物品全部放進無菌室,檢驗過程中嚴禁進出實驗室取用實驗用品。在無菌室內檢驗用具要保存在固定位置,杜絕隨意移動,無關物品禁止入內。生物安全櫃應保持台麵清潔,使用前按規定進行消毒,還要定期委托有資質的第三方實驗室進行校準,對濾網等關鍵配件要定期清理或更換。實驗中人員是潔淨室內主要的發塵源,單位麵積內人員越多,對實驗環境影響越大,實驗結果的可信度越差。因此,實驗時無關人員禁止入內,同時檢驗操作應平穩有序,嚴禁人員頻繁走動、接打電話,以避免攪動空氣中的塵埃粒子,影響檢驗結果。
3.5 菌落計數
菌落計數方法首先要考慮方法的選擇及確認。一般首選操作過程簡單的方法,常用的是低稀釋級(1∶10稀釋液)平皿傾注法,從而減少操作繁雜帶來誤差或者損傷微生物。其次選用增加稀釋倍數或培養基體積、添加中和劑或滅活劑等去除或滅活供試液的抗菌活性,再次選擇薄膜過濾法,或者上述幾種方法聯用。最可能數法因為精密度及準確度較差一般不選用。無論采取哪種方法,均要考慮所添加的表麵活性劑、中和劑、滅活劑等試劑對微生物無毒性,同時確認所用試劑的相容性,避免發生反應影響檢驗結果。實驗中一定要進行陰性對照試驗,在出現菌落生長時,便於區分藥品自身情況還是操作環境等其他因素影響。
菌落計數時一般通過直接觀察,在平皿的背麵進行菌落計數,計數時應注意平皿邊緣生長的菌落,同時注意區分菌落和顆粒物、膠囊殼碎片等,可借助放大鏡、低倍顯微鏡觀察,必要時挑取可疑物進行塗片鏡檢。
藥典中規定的有需氧菌總數、黴菌和酵母菌總數、控製菌檢查。微生物廣泛存在於自然界中,藥品中汙染微生物的種類有可能不在其規定範圍內。出現不明菌落時,應進行鑒定,確定汙染的原因並及時采取預防措施。一旦確定是樣品自身原因,應立即上報監管部門,避免發生藥害事件。
3.6 藥品自身性質
合格的藥品是生產出來的,不是檢驗出來的。在藥物生產、運輸、存儲過程中,都有可能會汙染微生物。藥品的生產環境、工藝、設備、原料及輔料、質量控製方法及水平和生產人員等均有可能帶來藥品微生物的汙染。
實驗室收到樣品後,首先應做好唯一性標識,避免樣品混淆,尤其針對三同樣品(同品種同廠家同批號)。應嚴格按照說明書上記載的貯藏方式保存樣品,最大限度保持樣品原始狀態,避免樣品貯藏條件比如相對濕度過高、溫度過高等不符合要求導致的微生物汙染。
藥品中可能含有防腐劑,如果防腐劑未被中和或滅活,平板計數結果往往會受到影響,如低稀釋級菌落生長數量較少,而高稀釋級菌落生長數量反而增大,遇此情況應重複試驗,確定是藥品自身性質影響還是操作技術造成的誤差。樣品的給藥途徑、抑菌程度等因素會影響檢驗方法的選擇,不同的檢驗方法又可能帶來不同的誤差來源。
4 討論及改進措施
針對誤差因素的分析,有條件的實驗室應配備微生物及相關專業畢業、有較高專業知識水平的人員。檢驗人員上崗前,必須進行嚴格的崗前培訓,要熟練操作儀器設備,明確無菌操作要求,按規定製備培養基、潔淨室的消毒、培養基及廢棄物的滅菌、平板製作、菌落的計數方法、菌落的鑒定、菌種的保藏及傳代、潔淨室的微生物監測等。要對檢驗人員加強生物安全培訓,熟悉安全操作規範和滅菌消毒知識,保證自身安全的同時,也能有效防止實驗室被微生物汙染。
出現不合格結果時,應及時啟動偏差調查,評估是樣品的原因還是實驗室誤差。積極參加外部組織的能力驗證等方式外,可在實驗室內部開展盲樣檢測等活動來考察檢驗技能和水平。當開展新項目、使用新方法或新技術時,檢測開始前對其操作技能要進行專項評估確認。
藥典中的微限檢查對象是活的微生物,而藥品中汙染的微生物具有分布不均勻性。同一批次藥品可能部分被汙染;被汙染的藥品中,汙染微生物的數量可能也不一致;被汙染藥品微生物種類可能比較複雜,也可能比較單一。微生物具有族團性,其大小及緊密程度具有遺傳性,族團的分化性差異極大,進一步強化分布不均勻性。針對上述特點,抽樣時應嚴格遵守抽樣指導原則,保證樣品具有代表性。
要強化檢驗人員對微限檢查全過程的質量控製意識,做到以藥典指導原則為綱,落到細處,落到實處。建立本實驗室微限檢查的標準操作規程,使微限檢查逐步趨向規範化,標準化,既要防止假陽性結果,更要避免假陰性結果,應客觀反映出藥品中微生物汙染程度,確保檢驗結果的準確性,為藥品安全有效提供技術支撐。
來源:菏澤市食品藥品檢驗檢測研究院 曹魯娜 畢言鋒 任仲麗 畢天琛 李懷偉
提醒:本文章所有內容均來源網絡,僅用於學習交流,若有侵權內容,請及時聯係刪除或修改,特此聲明!
上一篇:基因突變的類型
下一篇:微生物培養基主要成分綜述


